数据与计算发展前沿 ›› 2023, Vol. 5 ›› Issue (4): 27-37.
CSTR: 32002.14.jfdc.CN10-1649/TP.2023.04.003
doi: 10.11871/jfdc.issn.2096-742X.2023.04.003
陈美霖1,2( ),刘端阳3,徐黎明1,2,汪洋1,*()
),刘端阳3,徐黎明1,2,汪洋1,*()
收稿日期:2023-06-01
出版日期:2023-08-20
发布日期:2023-08-23
通讯作者:
*汪洋(E-mail: 作者简介:陈美霖,中国科学院计算机网络信息中心,硕士研究生,主要研究方向为机器学习力场等。基金资助:
CHEN Meilin1,2(),LIU Duanyang3,XU Liming1,2,WANG Yang1,*()
Received:2023-06-01
Online:2023-08-20
Published:2023-08-23
摘要:
【应用背景】在过去的几十年里,由于原子结构以及计算的复杂性,传统力场方法在解决某些问题时较为吃力。【目的】而机器学习方法的引入,有望解决许多曾经无法攻克的难题,平衡计算效率和计算精度之间的制约关系。【方法】该方法不依赖于先入为主的知识,通过从小规模高精度分子动力学模拟数据中学习来对力场进行建模,同时对原子核和核外电子的运动做了近似假设,从而很大程度上简化了力场的生成过程。【结果】机器学习力场旨在达到与传统力场几乎同样的精度并大幅度地提高计算效率。本文概述了机器学习力场的发展以及其相关理论知识,介绍了几种比较常见的机器学习力场方法,最后探讨了机器学习力场的不足以及未来需要克服的挑战。
陈美霖, 刘端阳, 徐黎明, 汪洋. 基于机器学习的力场模型研究综述[J]. 数据与计算发展前沿, 2023, 5(4): 27-37.
CHEN Meilin, LIU Duanyang, XU Liming, WANG Yang. A Review of Force Field Models Based on Machine Learning[J]. Frontiers of Data and Computing, 2023, 5(4): 27-37, https://cstr.cn/32002.14.jfdc.CN10-1649/TP.2023.04.003.
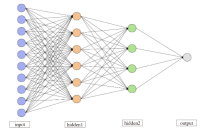
图1
多层神经网络结构"
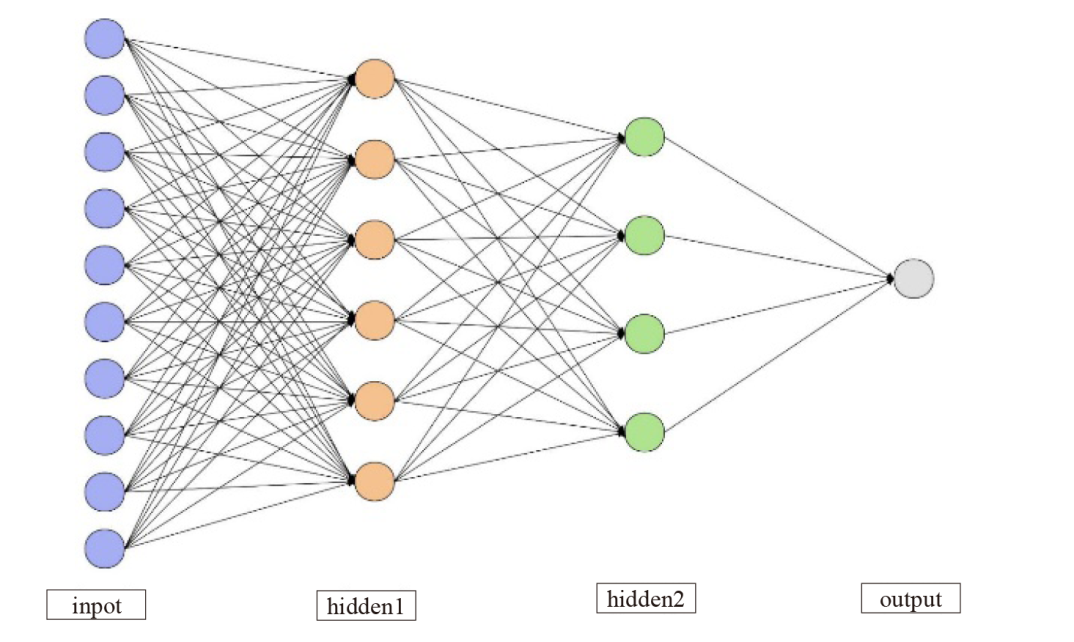
表1
GDML和sGDML的预测性能"
| GDML | sGDML | |||
|---|---|---|---|---|
| Force | Energy | Force | Energy | |
| Benzene | 0.195 | 0.07 | 0.16 | 0.07 |
| Uracil | 0.663 | 0.142 | 0.663 | 0.142 |
| Naphthalene | 0.222 | 0.12 | 0.113 | 0.116 |
| Aspirin | 0.984 | 0.264 | 0.679 | 0.194 |
| Salicylic Acid | 0.829 | 0.178 | 0.829 | 0.178 |
| Malonaldehyde | 0796 | 0.157 | 0.414 | 0.098 |
| Ethanol | 0.792 | 0.154 | 0.335 | 0.072 |
| Toluene | 0.425 | 0.125 | 0.142 | 0.097 |
| Paracetamol | 1.036 | 0.274 | 0.491 | 0.153 |
| Azobenzene | 0.78 | 0.353 | 0.409 | 0.092 |
表2
SchNet和sGDML的预测性能"
| SchNet | sGDML | |||
|---|---|---|---|---|
| Force | Energy | Force | Energy | |
| Benzene | 0.31 | 0.08 | 0.16 | 0.07 |
| Uracil | 0.56 | 0.17 | 0.663 | 0.142 |
| Naphthalene | 0.58 | 0.16 | 0.113 | 0.116 |
| Aspirin | 0.135 | 0.37 | 0.679 | 0.194 |
| Salicylic acid | 0.85 | 0.2 | 0.829 | 0.178 |
| Malonaldehyde | 0.66 | 0.13 | 0.414 | 0.098 |
| Ethanol | 0.39 | 0.08 | 0.335 | 0.072 |
| Toluene | 0.57 | 0.12 | 0.142 | 0.097 |
| [1] |
LIU D, XU L, LIN X, et al. Machine Learning for Semi-conductors[J]. Chip, 2022, 1(4): 100033.
doi: 10.1016/j.chip.2022.100033 |
| [2] |
FUKUHARA S, SHIMOJO F, SHIBUTA Y. Confor-mation and catalytic activity of nickel-carbon cluster for ethanol dissociation in carbon nanotube synthesis: Ab initio molecular dynamics simulation[J]. Chemical Physics Letters, 2017, 679: 164-171.
doi: 10.1016/j.cplett.2017.04.086 |
| [3] |
CHMIELA S, SAUCEDA H E, MÜLLER K R, et al. Tow-ards exact molecular dynamics simulations with machine-learned force fields[J]. Nature communications, 2018, 9(1): 3887.
doi: 10.1038/s41467-018-06169-2 |
| [4] |
TUCKERMAN M E. molecular dynamics: basic conc-epts, current trends and novel applications[J]. Journal of Physics Condensed Matter, 14(50): R1297-R1355.
doi: 10.1088/0953-8984/14/50/202 |
| [5] | CHU Z M, XIAO D J, QIAO Y S, et al. Machine Learn-ing Applications for Particle Accelerators[J]. Frontiers of Data and Computing, 2019, 1(2): 110-120. |
| [6] |
UNKE O T, CHMIELA S, SAUCEDA H E, et al. Mach-ine learning force fields[J]. Chemical Reviews, 2021, 121(16): 10142-10186.
doi: 10.1021/acs.chemrev.0c01111 |
| [7] |
BARTÓK A P, PAYNE M C, KONDOR R, et al. Gaus-sian approximation potentials: The accuracy of quantum mechanics, without the electrons[J]. Physical review letters, 2010, 104(13): 136403.
doi: 10.1103/PhysRevLett.104.136403 |
| [8] | UNKE O T, MEUWLY M. PhysNet: A neural network for predicting energies, forces, dipole moments, and partial charges[J]. Journal of chemical theory and compu-tation, 2019, 15(6): 3678-3693. |
| [9] |
BEHLER J, PARRINELLO M. Generalized neural-network representation of high-dimensional potential-energy surfaces[J]. Physical review letters, 2007, 98(14): 146401.
doi: 10.1103/PhysRevLett.98.146401 |
| [10] |
ZHANG L, HAN J, WANG H, et al. Deep potential molecular dynamics: a scalable model with the accuracy of quantum mechanics[J]. Physical review letters, 2018, 120(14): 143001.
doi: 10.1103/PhysRevLett.120.143001 |
| [11] |
STÖHR M, MEDRANO SANDONAS L, TKATC-HENKO A. Accurate many-body repulsive potentials for density-functional tight binding from deep tensor neural networks[J]. The Journal of Physical Chemistry Letters, 2020, 11(16): 6835-6843.
doi: 10.1021/acs.jpclett.0c01307 |
| [12] | CHMIELA S, VASSILEV-GALINDO V, UNKE O T, et al. Accurate global machine learning force fields for molecules with hundreds of atoms[J]. Science Advances, 2023, 9(2): eadf0873. |
| [13] | GUO J L, WANG Z G, WANG Y G, et al. A Review of Material Research and Development Methods Based on Computer Technology[J]. Frontiers of Data and Comp-uting, 2021, 3(2): 120-132. |
| [14] | WANG Z G, WAN M, CHEN Z Y, et al. Research and Application of a Data-Driven Intelligent Design Platform for Materials[J]. Frontiers of Data and Computing, 2023, 5(2): 86-96. |
| [15] |
SZE V, CHEN Y H, YANG T J, et al. Efficient processing of deep neural networks: A tutorial and survey[J]. Proceedings of the IEEE, 2017, 105(12): 2295-2329.
doi: 10.1109/JPROC.2017.2761740 |
| [16] |
BARTÓK A P, KERMODE J, BERNSTEIN N, et al. Machine learning a general-purpose interatomic potential for silicon[J]. Physical Review X, 2018, 8(4): 041048.
doi: 10.1103/PhysRevX.8.041048 |
| [17] |
SAUCEDA H E, VASSILEV-GALINDO V, CHMIELA S, et al. Dynamical strengthening of covalent and non-covalent molecular interactions by nuclear quantum effe-cts at finite temperature[J]. Nature Communications, 2021, 12(1): 442.
doi: 10.1038/s41467-020-20212-1 |
| [18] |
SCHÜTT K T, ARBABZADAH F, CHMIELA S, et al. Quantum-chemical insights from deep tensor neural networks[J]. Nature communications, 2017, 8(1): 13890.
doi: 10.1038/ncomms13890 |
| [19] | 申林, 贾璐阳, 汤典东, 等. 计算化学中的机器学习[J]. 中国科学:化学, 2022, 52(6): 858-868 |
| [20] |
POLTAVSKY I, TKATCHENKO A. Machine learn-ing force fields: Recent advances and remaining challe-nges[J]. The Journal of Physical Chemistry Letters, 2021, 12(28): 6551-6564.
doi: 10.1021/acs.jpclett.1c01204 |
| [21] |
WEI J, CHU X, SUN X Y, et al. Machine learning in materials science[J]. InfoMat, 2019, 1(3): 338-358.
doi: 10.1002/inf2.v1.3 |
| [22] | TU Y Y, ZHENG Q J, ZHAO J. Research on Quantum Proton Coupled Charge Transfer Process Based on Deep Neural Network[J]. Frontiers of Data and Computing, 2023, 5(2): 37-49. |
| [23] |
ROSENBERGER D, SMITH J S, GARCIA A E. Mod-eling of peptides with classical and novel machine learning force fields: A comparison[J]. The Journal of Physical Chemistry B, 2021, 125(14): 3598-3612.
doi: 10.1021/acs.jpcb.0c10401 |
| [24] |
SMITH J S, NEBGEN B T, ZUBATYUK R, et al. Approaching coupled cluster accuracy with a general-purpose neural network potential through transfer lear-ning[J]. Nature communications, 2019, 10(1): 2903.
doi: 10.1038/s41467-019-10827-4 |
| [25] |
SMITH J S, ISAYEV O, ROITBERG A E. ANI-1: an extensible neural network potential with DFT accuracy at force field computational cost[J]. Chemical science, 2017, 8(4): 3192-3203.
doi: 10.1039/c6sc05720a pmid: 28507695 |
| [26] |
SMITH J S, ZUBATYUK R, NEBGEN B, et al. The ANI-1ccx and ANI-1x data sets, coupled-cluster and density functional theory properties for molecules[J]. Scientific data, 2020, 7(1): 134.
doi: 10.1038/s41597-020-0473-z pmid: 32358545 |
| [27] | HU W, SHUAIBI M, DAS A, et al. Forcenet: A graph neural network for large-scale quantum calculations[J]. arXiv preprint arXiv:2103.01436, 2021. |
| [28] | CHMIELA S, TKATCHENKO A, SAUCEDA H E, et al. Machine learning of accurate energy-conserving molecular force fields[J]. Science advances, 2017, 3(5): e1603015. |
| [29] |
CHMIELA S, SAUCEDA H E, POLTAVSKY I, et al. sGDML: Constructing accurate and data efficient mole-cular force fields using machine learning[J]. Computer Physics Communications, 2019, 240: 38-45.
doi: 10.1016/j.cpc.2019.02.007 |
| [30] | GASTEIGER J, GROß J, GÜNNEMANN S. Directional message passing for molecular graphs[J]. arXiv preprint arXiv:2003.03123, 2020. |
| [31] | GASTEIGER J, GIRI S, MARGRAF J T, et al. Fast and uncertainty-aware directional message passing for non-equilibrium molecules[J]. arXiv preprint arXiv: 2011.14115, 2020. |
| [32] |
SCHÜTT K T, HESSMANN S S P, GEBAUER N W A, et al. SchNetPack 2.0: A neural network toolbox for atomistic machine learning[J]. The Journal of Chemical Physics, 2023, 158(14): 144801.
doi: 10.1063/5.0138367 |
| [33] | SCHÜTT K, KINDERMANS P J, SAUCEDA FELIX H E, et al. Schnet: A continuous-filter convolutional neural network for modeling quantum interactions[J]. Advances in neural information processing systems, 2017, 30: 991-1001. |
| [34] | ZENI C, ROSSI K, GLIELMO A, et al. On machine learning force fields for metallic nanoparticles[J]. Adv-ances in Physics: X, 2019, 4(1): 1654919. |
| [35] |
ZHANG W, WENG M, ZHANG M, et al. Revealing Morphology Evolution of Lithium Dendrites by Large-Scale Simulation Based on Machine Learning Force Field[J]. Advanced Energy Materials, 2023, 13(4): 2202892.
doi: 10.1002/aenm.v13.4 |
| [36] |
LI Z, MEIDANI K, YADAV P, et al. Graph neural net-works accelerated molecular dynamics[J]. The Journal of Chemical Physics, 2022, 156(14): 144103.
doi: 10.1063/5.0083060 |
| [37] | KABYLDA A, VASSILEV-GALINDO V, CHMIELA S, et al. Towards linearly scaling and chemically accurate global machine learning force fields for large molecules[J]. arXiv preprint arXiv:2209.03985, 2022. |
| [38] | SAUCEDA H E, GÁLVEZ-GONZÁLEZ L E, CHM-IELA S, et al. BIGDML: Towards Exact Machine Lear-ning Force Fields for Materials[J]. arXiv e-prints, 2021: arXiv: 2106.04229. |
| [39] |
GASTEGGER M, SCHWIEDRZIK L, BITTERMANN M, et al. wACSF—Weighted atom-centered symmetry functions as descriptors in machine learning potentials[J]. The Journal of chemical physics, 2018, 148(24): 241709.
doi: 10.1063/1.5019667 |
| [40] |
LUBBERS N, SMITH J S, BARROS K. Hierarchical modeling of molecular energies using a deep neural network[J]. The Journal of chemical physics, 2018, 148(24): 241715.
doi: 10.1063/1.5011181 |
| [41] |
SCHÜTT K T, KESSEL P, GASTEGGER M, et al. Sch-NetPack: A deep learning toolbox for atomistic syste-ms[J]. Journal of chemical theory and computation, 2018, 15(1): 448-455.
doi: 10.1021/acs.jctc.8b00908 |
| [42] |
SCHÜTT K T, SAUCEDA H E, KINDERMANS P J, et al. Schnet-a deep learning architecture for molecules and materials[J]. The Journal of Chemical Physics, 2018, 148 (24): 241722.
doi: 10.1063/1.5019779 |
| [43] | BATTAGLIA P, PASCANU R, LAI M, et al. Interaction networks for learning about objects, relations and physics[J]. Advances in neural information processing systems, 2016, 29: 4509-4517. |
| [44] |
XIE T, GROSSMAN J C. Crystal graph convolutional neural networks for an accurate and interpretable pre-diction of material properties[J]. Physical review letters, 2018, 120(14): 145301.
doi: 10.1103/PhysRevLett.120.145301 |
| [45] | FU X, WU Z, WANG W, et al. Forces are not enough: Benchmark and critical evaluation for machine learning force fields with molecular simulations[J]. arXiv preprint arXiv:2210.07237, 2022. |
| [46] |
GKEKA P, STOLTZ G, BARATI FARIMANI A, et al. Machine learning force fields and coarse-grained variables in molecular dynamics: application to materials and biological systems[J]. Journal of chemical theory and computation, 2020, 16(8): 4757-4775.
doi: 10.1021/acs.jctc.0c00355 pmid: 32559068 |
| [47] |
MISHIN Y. Machine-learning interatomic potentials for materials science[J]. Acta Materialia, 2021, 214: 116980.
doi: 10.1016/j.actamat.2021.116980 |
| [48] |
SCHMIDT J, MARQUES M R G, BOTTI S, et al. Recent advances and applications of machine learning in solid-state materials science[J]. Npj Computational Materials, 2019, 5(1): 83.
doi: 10.1038/s41524-019-0221-0 |
| [49] |
MILARDOVICH D, WALDHOER D, JECH M, et al. Building robust machine learning force fields by composite Gaussian approximation potentials[J]. Solid-State Electronics, 2023, 200: 108529.
doi: 10.1016/j.sse.2022.108529 |
| [50] |
SCHMITZ N F, MÜLLER K R, CHMIELA S. Alg-orithmic differentiation for automated modeling of machine learned force fields[J]. The Journal of Physical Chemistry Letters, 2022, 13(43): 10183-10189.
doi: 10.1021/acs.jpclett.2c02632 |
| [51] |
LIAO K, DONG S, CHENG Z, et al. Combined frag-ment-based machine learning force field with classical force field and its application in the NMR calculations of macromolecules in solutions[J]. Physical Chemistry Chemical Physics, 2022, 24(31): 18559-18567.
doi: 10.1039/D2CP02192G |
| [1] | 孟哲, 余粟. 基于Spark和优化BP神经网络的出租车需求预测模型[J]. 数据与计算发展前沿, 2023, 5(4): 112-126. |
| [2] | 田一擎, 程曦, 冯博靖. 企业信用评级计算模型综述[J]. 数据与计算发展前沿, 2023, 5(4): 139-153. |
| [3] | 刘端阳, 魏钟鸣. 有监督学习算法在材料科学中的应用[J]. 数据与计算发展前沿, 2023, 5(4): 38-47. |
| [4] | 陈栋, 李明, 陈淑文. 结合Transformer和多层特征聚合的高光谱图像分类算法[J]. 数据与计算发展前沿, 2023, 5(3): 138-151. |
| [5] | 许淞源,刘峰. ESDRec:一种面向地球大数据平台的数据推荐模型[J]. 数据与计算发展前沿, 2023, 5(1): 55-64. |
| [6] | 童昭,王露笛,朱小杰,杜一. 基于预训练模型的军事领域命名实体识别研究[J]. 数据与计算发展前沿, 2022, 4(5): 120-128. |
| [7] | 石雪梅,朱克亮,张祥民,张树涛,陈良锋. 基于生成对抗网络的有遮挡人脸修复方法[J]. 数据与计算发展前沿, 2022, 4(4): 123-131. |
| [8] | 肖楠,周明珠,邢军,罗泽,李晓辉. 基于高分辨率网络和注意力机制的真伪卷烟包装鉴别[J]. 数据与计算发展前沿, 2021, 3(5): 118-129. |
| [9] | 张猛,李健. 鸟类音频数据预处理方法[J]. 数据与计算发展前沿, 2021, 3(5): 130-140. |
| [10] | 张晨阳,杜义华. 短文本自动生成技术研究进展[J]. 数据与计算发展前沿, 2021, 3(3): 111-125. |
| [11] | 陈子健,李俊,岳兆娟,赵泽方. 基于自编码器与属性信息的混合推荐模型[J]. 数据与计算发展前沿, 2021, 3(3): 148-155. |
| [12] | 陈涛,安俊秀. 基于特征融合的微博短文本情感分类研究[J]. 数据与计算发展前沿, 2020, 2(6): 21-29. |
| [13] | 刘晓东,倪浩然. 深度学习技术在学科融合研究中的应用[J]. 数据与计算发展前沿, 2020, 2(5): 99-109. |
| [14] | 杨健鹏,罗泽,张应明. 土地利用演变过程的人工智能建模[J]. 数据与计算发展前沿, 2020, 2(3): 137-145. |
| [15] | 欧阳与点,谢鲲. 网络性能数据恢复算法[J]. 数据与计算发展前沿, 2020, 2(3): 55-65. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||