数据与计算发展前沿 ›› 2026, Vol. 8 ›› Issue (2): 184-203.
CSTR: 32002.14.jfdc.CN10-1649/TP.2026.02.014
doi: 10.11871/jfdc.issn.2096-742X.2026.02.014
许黄超1,2( ),张宝花1,*(),刘倩1,金钟1,*()
),张宝花1,*(),刘倩1,金钟1,*()
收稿日期:2025-08-08
出版日期:2026-04-20
发布日期:2026-04-23
通讯作者:
*张宝花(E-mail: 作者简介:许黄超,中国科学院计算机网络信息中心,中国科学院大学,博士研究生,主要研究方向为人工智能辅助药物设计。基金资助:
XU Huangchao1,2(),ZHANG Baohua1,*(),LIU Qian1,JIN Zhong1,*()
Received:2025-08-08
Online:2026-04-20
Published:2026-04-23
摘要:
【目的】 在人工智能(AI)技术与海量分子数据的双重驱动下,AI赋能的分子生成已成为药物设计与化学创新的关键技术。本文聚焦于小分子设计,旨在系统综述AI驱动的小分子生成方法及在药物研发中的应用。【文献范围】梳理了国内外支持小分子生成的主要数据资源、生成方法与应用研究。【方法】 围绕变分自编码器、生成对抗网络、Transformer、扩散模型及大语言模型等技术路线,介绍当前主流模型及其核心机制,结合靶点引导、结构约束与语言建模等策略展开归纳。【结果】 AI小分子生成方法在多个应用场景中展现出显著优势,但对数据质量、算法复杂度和计算资源提出更高要求。【局限】受篇幅限制,本文未能全面涵盖该领域的所有分支和最新进展。【结论】 AI驱动的小分子生成方法正在加速分子发现和药物设计创新进程,构建AI-ready的高质量数据集,提升小分子生成模型的可控性和泛化能力,完善生成评价体系将是未来重要的研究方向。
许黄超, 张宝花, 刘倩, 金钟. 人工智能驱动的分子生成方法与数据资源综述[J]. 数据与计算发展前沿, 2026, 8(2): 184-203.
XU Huangchao, ZHANG Baohua, LIU Qian, JIN Zhong. A Review on AI-Driven Methods and Data Resources for Molecule Generation[J]. Frontiers of Data and Computing, 2026, 8(2): 184-203, https://cstr.cn/32002.14.jfdc.CN10-1649/TP.2026.02.014.

图1
AI发现分子进入临床试验的数量和类型统计"
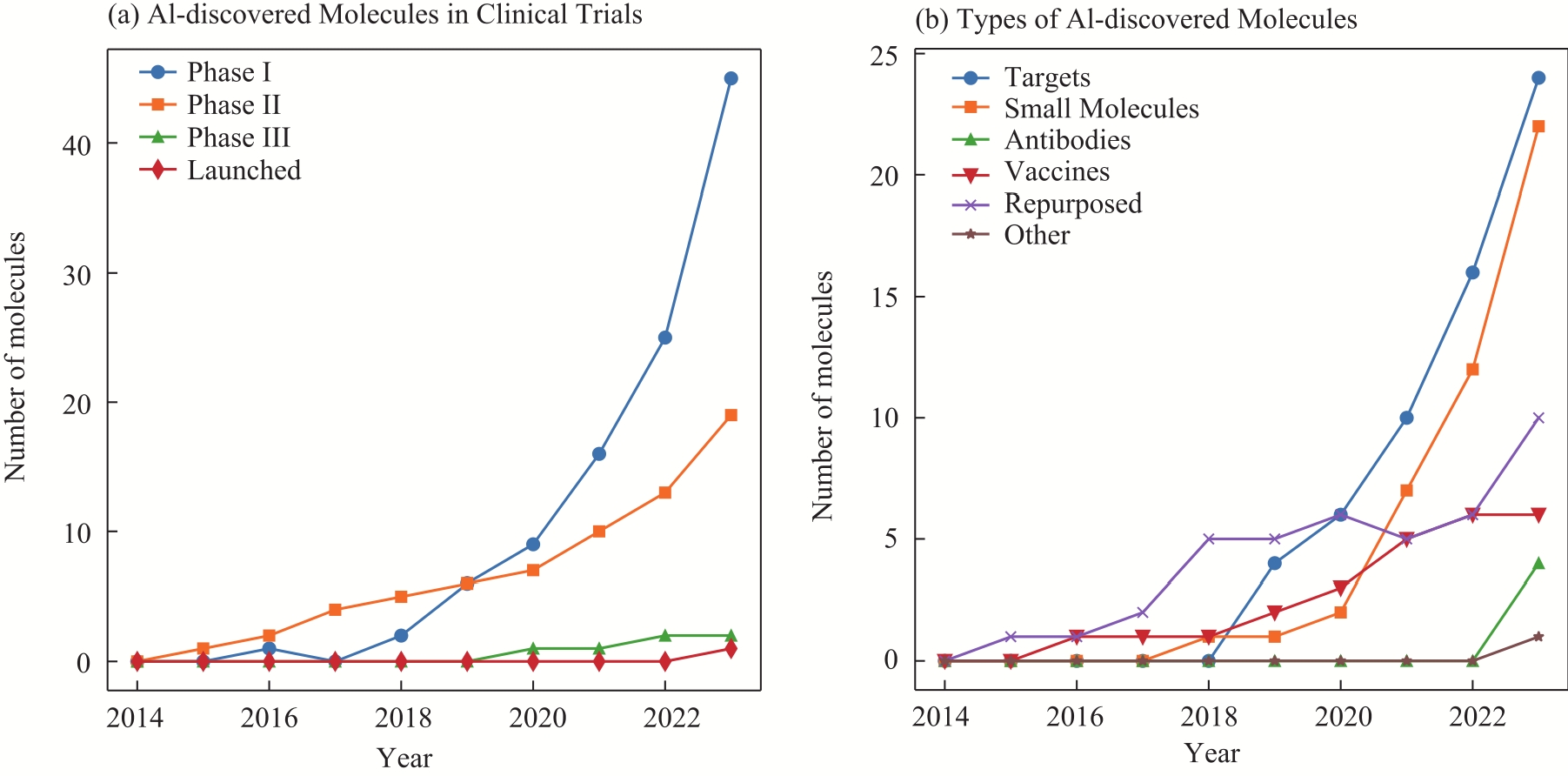
表1
分子生成相关数据集的统计细节"
| 分类 | 数据集 | 数据规模 | 原始数据格式 | 访问途径 |
|---|---|---|---|---|
| 小分子数据 | ZINC | 37 B | SMILES、SDF | |
| ChEMBL | 22.7 M | SMILES、InChI | | |
| PubChem | 750 M | SMILES、SDF | | |
| Enamine REAL | 10 B | SMILES、SDF | | |
| DrugBank | 1.5 M | SMILES、MOL、SDF | | |
| ChemDiv | 1.6 M | SMILES、SDF | | |
| 蛋白质和复合物数据 | RCSB PDB | 238 K | Experimental PDB | |
| AlphaFoldDB | 200 M | Predicted PDB | | |
| UniProt | 250 M | Sequence Entry | | |
| PDBBind | 35 K | PDB、SDF | | |
| Crossdocked | 22.5 M | PDB、SDF | | |
| KLIFS | 6 K | PDB、SDF | | |
| BindingDB | 1.2 M | SMILES、Sequence | | |
| 基准评估 数据 | MoleculeNet | 700 K | SMILES | |
| MOSES | 1.9 M | SMILES | | |
| GuacaMol | / | SMILES、MD5 | | |
| TDC | / | SMILES、PDB | | |
| ADMET Lab | 400 K | SMILES | |
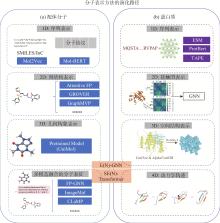
图2
配体分子和蛋白表示方法的演化路径"
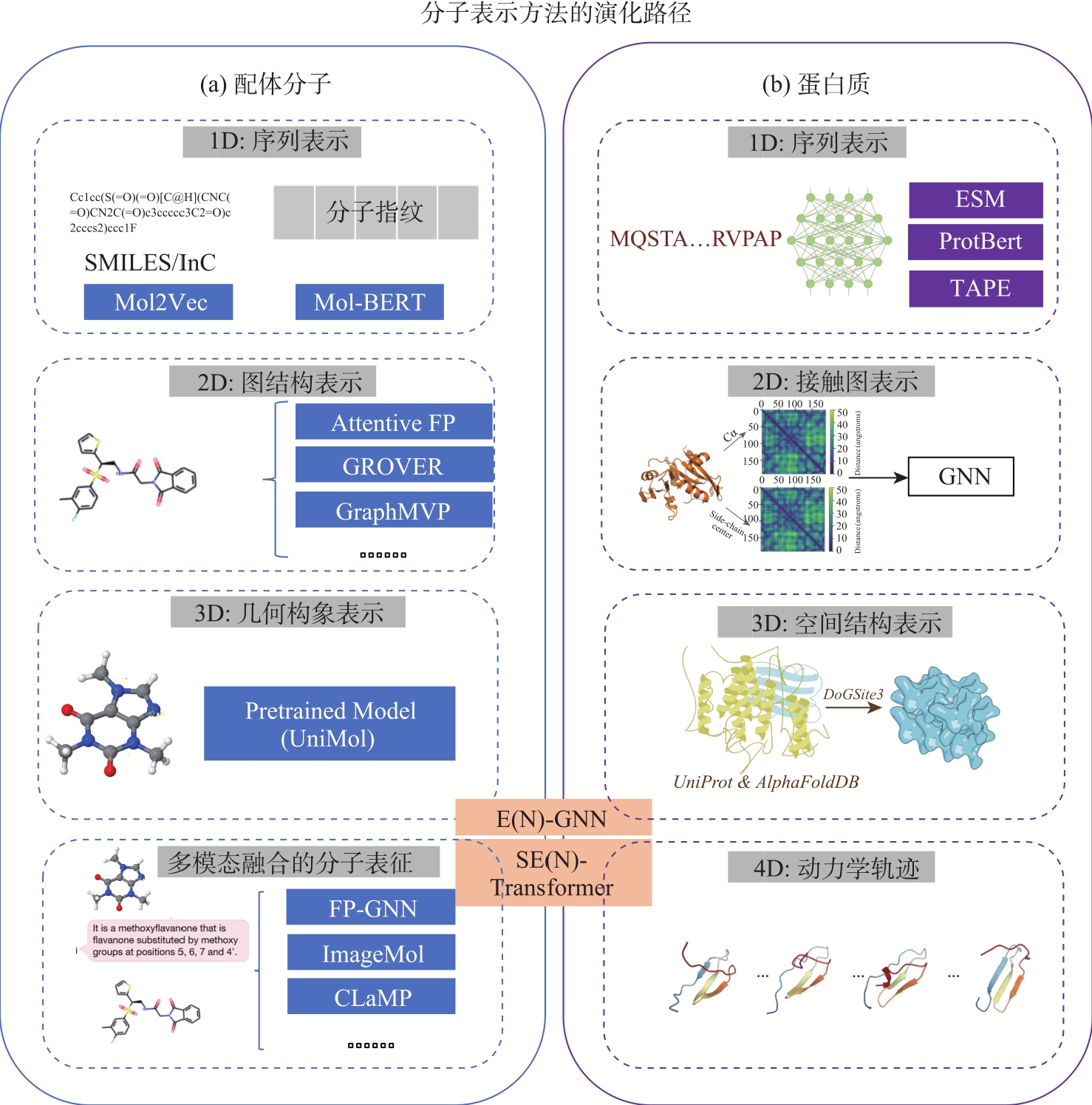
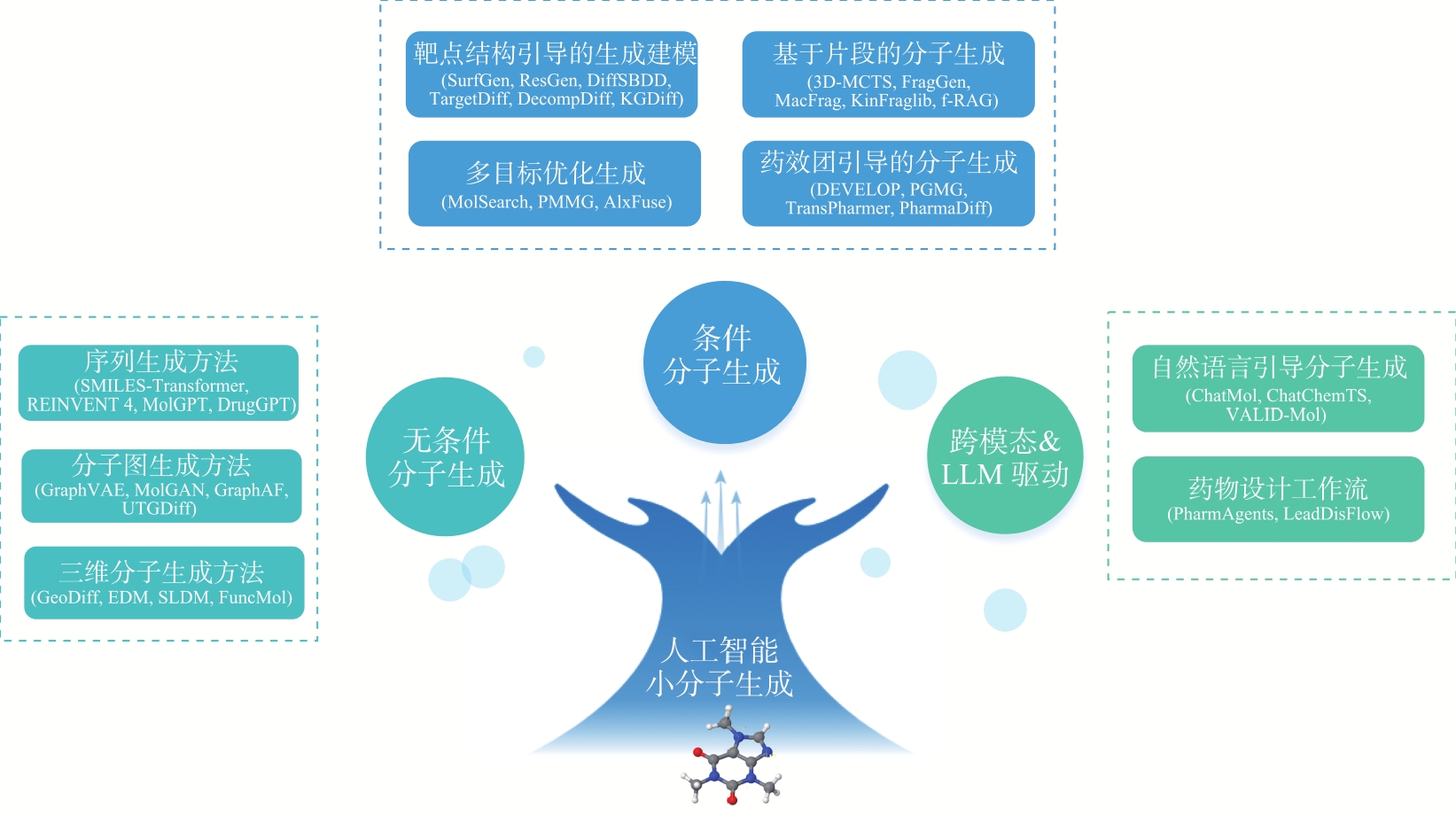
图3
本文综述的生成模型在药物发现中的应用分类"
| [1] |
JAYATUNGA M, AYERS M, BRUENS L, et al. How successful are AI-discovered drugs in clinical trials? A first analysis and emerging lessons[J]. Drug Discovery Today, 2024, 29: 104009.
doi: 10.1016/j.drudis.2024.104009 |
| [2] |
IRWIN J J, TANG K G, YOUNG J, et al. ZINC20—A Free Ultralarge-Scale Chemical Database for Ligand Discovery[J]. Journal of Chemical Information and Modeling, 2020, 60(12): 6065-6073.
doi: 10.1021/acs.jcim.0c00675 |
| [3] |
TINGLE B I, TANG K G, CASTANON M, et al. ZINC-22─A Free Multi-Billion-Scale Database of Tangible Compounds for Ligand Discovery[J]. Journal of Chemical Information and Modeling, 2023, 63(4): 1166-1176.
doi: 10.1021/acs.jcim.2c01253 pmid: 36790087 |
| [4] |
GAULTON A, BELLIS L J, BENTO A P, et al. ChEMBL: a large-scale bioactivity database for drug discovery[J]. Nucleic Acids Research, 2012, 40(Database issue): D1100-D1107.
doi: 10.1093/nar/gkr777 |
| [5] |
ZDRAZIL B, FELIX E, HUNTER F, et al. The ChEMBL Database in 2023: a drug discovery platform spanning multiple bioactivity data types and time periods[J]. Nucleic Acids Research, 2024, 52(D1): D1180-D1192.
doi: 10.1093/nar/gkad1004 |
| [6] |
KIM S, THIESSEN P A, BOLTON E E, et al. PubChem Substance and Compound databases[J]. Nucleic Acids Research, 2016, 44(Database issue): D1202-D1213.
doi: 10.1093/nar/gkv951 |
| [7] |
KIM S, CHEN J, CHENG T, et al. PubChem 2023 update[J]. Nucleic Acids Research, 2023, 51(D1): D1373-D1380.
doi: 10.1093/nar/gkac956 |
| [8] |
GRYGORENKO O O, RADCHENKO D S, DZIUBA I, et al. Generating Multibillion Chemical Space of Readily Accessible Screening Compounds[J]. iScience, 2020, 23(11): 101681.
doi: 10.1016/j.isci.2020.101681 |
| [9] |
WISHART D S, FEUNANG Y D, GUO A C, et al. DrugBank 5.0: a major update to the DrugBank database for 2018[J]. Nucleic Acids Research, 2018, 46(D1): D1074-D1082.
doi: 10.1093/nar/gkx1037 |
| [10] |
KNOX C, WILSON M, KLINGER C M, et al. DrugBank 6.0: the DrugBank Knowledgebase for 2024[J]. Nucleic Acids Research, 2024, 52(D1): D1265-D1275.
doi: 10.1093/nar/gkad976 |
| [11] |
SHANG J, SUN H, LIU H, et al. Comparative analyses of structural features and scaffold diversity for purchasable compound libraries[J]. Journal of Cheminformatics, 2017, 9(1): 25.
doi: 10.1186/s13321-017-0212-4 pmid: 29086044 |
| [12] |
BURLEY S K, BERMAN H M, BHIKADIYA C, et al. RCSB Protein Data Bank: biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy[J]. Nucleic Acids Research, 2019, 47(D1): D464-D474.
doi: 10.1093/nar/gky1004 |
| [13] |
UNIPROT CONSORTIUM T, BATEMAN A, MARTIN M J, et al. UniProt: the universal protein knowledgebase[J]. Nucleic Acids Research, 2017, 45(D1): D158-D169.
doi: 10.1093/nar/gkw1099 |
| [14] |
CONSORTIUM T U, BATEMAN A, MARTIN M J, et al. UniProt: the Universal Protein Knowledgebase in 2023[J]. Nucleic Acids Research, 2023, 51(D1): D523-D531.
doi: 10.1093/nar/gkac1052 |
| [15] |
JUMPER J, EVANS R, PRITZEL A, et al. Highly accurate protein structure prediction with AlphaFold[J]. Nature, 2021, 596(7873): 583-589.
doi: 10.1038/s41586-021-03819-2 |
| [16] |
ABRAMSON J, ADLER J, DUNGER J, et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3[J]. Nature, 2024, 630(8016): 493-500.
doi: 10.1038/s41586-024-07487-w |
| [17] |
WANG R, FANG X, LU Y, et al. The PDBbind database: methodologies and updates[J]. Journal of Medicinal Chemistry, 2005, 48(12): 4111-4119.
pmid: 15943484 |
| [18] |
LIU Z, LI Y, HAN L, et al. PDB-wide collection of binding data: current status of the PDBbind database[J]. Bioinformatics, 2015, 31(3): 405-412.
doi: 10.1093/bioinformatics/btu626 pmid: 25301850 |
| [19] |
FRANCOEUR P G, MASUDA T, SUNSERI J, et al. Three-Dimensional Convolutional Neural Networks and a Cross-Docked Data Set for Structure-Based Drug Design[J]. Journal of Chemical Information and Modeling, 2020, 60(9): 4200-4215.
doi: 10.1021/acs.jcim.0c00411 pmid: 32865404 |
| [20] |
VAN LINDEN O P J, KOOISTRA A J, LEURS R, et al. KLIFS: A Knowledge-Based Structural Database To Navigate Kinase-Ligand Interaction Space[J]. Journal of Medicinal Chemistry, 2014, 57(2): 249-277.
doi: 10.1021/jm400378w pmid: 23941661 |
| [21] |
KANEV G K, DE GRAAF C, WESTERMAN B A, et al. KLIFS: an overhaul after the first 5 years of supporting kinase research[J]. Nucleic Acids Research, 2020, 49(D1): D562-D569.
doi: 10.1093/nar/gkaa895 |
| [22] |
GILSON M K, LIU T, BAITALUK M, et al. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology[J]. Nucleic Acids Research, 2016, 44(Database issue): D1045-D1053.
doi: 10.1093/nar/gkv1072 |
| [23] |
LIU T, HWANG L, BURLEY S K, et al. BindingDB in 2024: a FAIR knowledgebase of protein-small molecule binding data[J]. Nucleic Acids Research, 2025, 53(D1): D1633-D1644.
doi: 10.1093/nar/gkae1075 pmid: 39574417 |
| [24] | WU Z, RAMSUNDAR B, FEINBERG E N, et al. MoleculeNet: A Benchmark for Molecular Machine Learning[Z]. arXiv, 2018[2025-07-21]. http://arxiv.org/abs/1703.00564. |
| [25] | POLYKOVSKIY D, ZHEBRAK A, SANCHEZ-LENGELING B, et al. Molecular Sets (MOSES): A Benchmarking Platform for Molecular Generation Models[Z]. arXiv, 2020[2025-07-10]. http://arxiv.org/abs/1811.12823. |
| [26] |
BROWN N, FISCATO M, SEGLER M H S, et al. GuacaMol: Benchmarking Models for de Novo Molecular Design[J]. Journal of Chemical Information and Modeling, 2019, 59(3): 1096-1108.
doi: 10.1021/acs.jcim.8b00839 pmid: 30887799 |
| [27] | HUANG K, FU T, GAO W, et al. Therapeutics Data Commons: Machine Learning Datasets and Tasks for Drug Discovery and Development[Z]. arXiv, 2021[2025-07-10]. http://arxiv.org/abs/2102.09548. |
| [28] |
DONG J, WANG N N, YAO Z J, et al. ADMETlab: a platform for systematic ADMET evaluation based on a comprehensively collected ADMET database[J]. Journal of Cheminformatics, 2018, 10(1): 29.
doi: 10.1186/s13321-018-0283-x pmid: 29943074 |
| [29] |
XIONG G, WU Z, YI J, et al. ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties[J]. Nucleic Acids Research, 2021, 49(W1): W5-W14.
doi: 10.1093/nar/gkab255 pmid: 33893803 |
| [30] |
FU L, SHI S, YI J, et al. ADMETlab 3.0: an updated comprehensive online ADMET prediction platform enhanced with broader coverage, improved performance, API functionality and decision support[J]. Nucleic Acids Research, 2024, 52(W1): W422-W431.
doi: 10.1093/nar/gkae236 |
| [31] |
ROGERS D, HAHN M. Extended-Connectivity Fingerprints[J]. Journal of Chemical Information and Modeling, 2010, 50(5): 742-754.
doi: 10.1021/ci100050t pmid: 20426451 |
| [32] |
KUWAHARA H, GAO X. Analysis of the effects of related fingerprints on molecular similarity using an eigenvalue entropy approach[J]. Journal of Cheminformatics, 2021, 13(1): 27.
doi: 10.1186/s13321-021-00506-2 pmid: 33757582 |
| [33] |
JAEGER S, FULLE S, TURK S. Mol2vec: unsupervised machine learning approach with chemical intuition[J]. Journal of chemical information and modeling, 2018, 58(1): 27-35.
doi: 10.1021/acs.jcim.7b00616 pmid: 29268609 |
| [34] | FABIAN B, EDLICH T, GASPAR H, et al. Molecular representation learning with language models and domain-relevant auxiliary tasks[Z]. arXiv, 2020[2025-07-21]. http://arxiv.org/abs/2011.13230. |
| [35] |
XIONG Z, WANG D, LIU X, et al. Pushing the Boundaries of Molecular Representation for Drug Discovery with the Graph Attention Mechanism[J]. Journal of Medicinal Chemistry, 2020, 63(16): 8749-8760.
doi: 10.1021/acs.jmedchem.9b00959 pmid: 31408336 |
| [36] | RONG Y, BIAN Y, XU T, et al. Self-Supervised Graph Transformer on Large-Scale Molecular Data[Z]. arXiv, 2020[2025-07-21]. http://arxiv.org/abs/2007.02835. |
| [37] | LIU S, WANG H, LIU W, et al. Pre-training Molecular Graph Representation with 3D Geometry[Z]. arXiv, 2022[2025-07-21]. http://arxiv.org/abs/2110.07728. |
| [38] | BATZNER S, MUSAELIAN A, SUN L, et al. E(3)-Equivariant Graph Neural Networks for Data-Efficient and Accurate Interatomic Potentials[J/OL]. Nature Communications, 2022, 13(1)[2025-07-10]. http://arxiv.org/abs/2101.03164. |
| [39] | FUCHS F B, WORRALL D E, FISCHER V, et al. SE(3)-Transformers: 3D Roto-Translation Equivariant Attention Networks[Z]. arXiv, 2020[2025-07-10]. http://arxiv.org/abs/2006.10503. |
| [40] |
LU S, GAO Z, HE D, et al. Data-driven quantum chemical property prediction leveraging 3D conformations with Uni-Mol+[J]. Nature Communications, 2024, 15(1): 7104.
doi: 10.1038/s41467-024-51321-w pmid: 39160169 |
| [41] | CAI H, ZHANG H, ZHAO D, et al. FP-GNN: a versatile deep learning architecture for enhanced molecular property prediction[J]. Briefings in Bioinformatics, 2022, 23(6): bbac408. |
| [42] | WU S, YU D, TAN X, et al. CLaMP: Contrastive Language-Music Pre-training for Cross-Modal Symbolic Music Information Retrieval[Z]. arXiv, 2023[2025-07-21]. http://arxiv.org/abs/2304.11029. |
| [43] | KAUFMAN B, WILLIAMS E, UNDERKOFFLER C, et al. COATI: multi-modal contrastive pre-training for representing and traversing chemical space[Z]. ChemRxiv, 2023[2025-07-21]. https://chemrxiv.org/engage/chemrxiv/article-details/64e8137fdd1a73847f73f7aa. |
| [44] | LIN Z, AKIN H, RAO R, et al. Evolutionary-scale prediction of atomic level protein structure with a language model[Z]. bioRxiv, 2022: 2022.07.20.500902[2025-07-10]. https://www.biorxiv.org/content/10.1101/2022.07.20.500902v3. |
| [45] | HAYES T, RAO R, AKIN H, et al. Simulating 500 million years of evolution with a language model[Z]. bioRxiv, 2024: 2024.07.01.600583[2025-07-10]. https://www.biorxiv.org/content/10.1101/2024.07.01.600583v1. |
| [46] |
BRANDES N, OFER D, PELEG Y, et al. ProteinBERT: a universal deep-learning model of protein sequence and function[J]. Bioinformatics, 2022, 38(8): 2102-2110.
doi: 10.1093/bioinformatics/btac020 pmid: 35020807 |
| [47] | RAO R, BHATTACHARYA N, THOMAS N, et al. Evaluating Protein Transfer Learning with TAPE[Z]. arXiv, 2019[2025-07-10]. http://arxiv.org/abs/1906.08230. |
| [48] |
JIANG M, LI Z, ZHANG S, et al. Dru-target affinity prediction using graph neural network and contact maps[J]. RSC Advances, 2020, 10(35): 20701-20712.
doi: 10.1039/D0RA02297G |
| [49] |
KLOCZKOWSKI A, JERNIGAN R L, WU Z, et al. Distance Matrix-Based Approach to Protein Structure Prediction[J]. Journal of structural and functional genomics, 2009, 10(1): 67-81.
doi: 10.1007/s10969-009-9062-2 pmid: 19224393 |
| [50] | ZHAO L, WANG H, SHI S. PocketDTA: an advanced multimodal architecture for enhanced prediction of drug-target affinity from 3D structural data of target binding pockets[J]. Bioinformatics, 2024, 40(10): btae594. |
| [51] | JING B, EISMANN S, SURIANA P, et al. Learning from Protein Structure with Geometric Vector Perceptrons[Z]. arXiv, 2021[2025-07-10]. http://arxiv.org/abs/2009.01411. |
| [52] | LIU C, WANG J, CAI Z, et al. Dynamic PDB: A New Dataset and a SE(3) Model Extension by Integrating Dynamic Behaviors and Physical Properties in Protein Structures[Z]. arXiv, 2024[2025-07-21]. http://arxiv.org/abs/2408.12413. |
| [53] | JIN W, BARZILAY R, JAAKKOLA T. Junction Tree Variational Autoencoder for Molecular Graph Generation[Z]. arXiv, 2019[2025-07-10]. http://arxiv.org/abs/1802.04364. |
| [54] | KUSNER M J, PAIGE B, HERNÁNDEZ-LOBATO J M. Grammar Variational Autoencoder[C]// Proceedings of the 34th International Conference on Machine Learning. PMLR, 2017: 1945-1954. |
| [55] | CAO N D, KIPF T. MolGAN: An implicit generative model for small molecular graphs[Z]. arXiv, 2022[2025-07-21]. http://arxiv.org/abs/1805.11973. |
| [56] | GUIMARAES G L, SANCHEZ-LENGELING B, OUTEIRAL C, et al. Objective-Reinforced Generative Adversarial Networks (ORGAN) for Sequence Generation Models[Z]. arXiv, 2018[2025-07-10]. http://arxiv.org/abs/1705.10843. |
| [57] | ZANG C, WANG F. MoFlow: An Invertible Flow Model for Generating Molecular Graphs[Z]. arXiv, 2020[2025-07-10]. http://arxiv.org/abs/2006.10137. |
| [58] | SONG Y, GONG J, XU M, et al. Equivariant Flow Matching with Hybrid Probability Transport[Z]. arXiv, 2023[2025-08-15]. http://arxiv.org/abs/2312.07168. |
| [59] | XU M, YU L, SONG Y, et al. GeoDiff: a Geometric Diffusion Model for Molecular Conformation Generation[Z]. arXiv, 2022[2025-07-10]. http://arxiv.org/abs/2203.02923. |
| [60] | HOOGEBOOM E, SATORRAS V G, VIGNAC C, et al. Equivariant Diffusion for Molecule Generation in 3D[Z]. arXiv, 2022[2025-07-10]. http://arxiv.org/abs/2203.17003. |
| [61] |
SHERSTINSKY A. Fundamentals of Recurrent Neural Network (RNN) and Long Short-Term Memory (LSTM) Network[J]. Physica D: Nonlinear Phenomena, 2020, 404: 132306.
doi: 10.1016/j.physd.2019.132306 |
| [62] | CHUNG J, GULCEHRE C, CHO K, et al. Empirical Evaluation of Gated Recurrent Neural Networks on Sequence Modeling[Z]. arXiv, 2014[2025-07-10]. http://arxiv.org/abs/1412.3555. |
| [63] |
BLASCHKE T, ARÚS-POUS J, CHEN H, et al. REINVENT 2.0: An AI Tool for De Novo Drug Design[J]. Journal of Chemical Information and Modeling, 2020, 60(12): 5918-5922.
doi: 10.1021/acs.jcim.0c00915 pmid: 33118816 |
| [64] |
LOEFFLER H H, HE J, TIBO A, et al. Reinvent 4: Modern AI-driven generative molecule design[J]. Journal of Cheminformatics, 2024, 16(1): 20.
doi: 10.1186/s13321-024-00812-5 pmid: 38383444 |
| [65] |
IRWIN R, DIMITRIADIS S, HE J, et al. Chemformer: a pre-trained transformer for computational chemistry[J]. Machine Learning: Science and Technology, 2022, 3(1): 015022.
doi: 10.1088/2632-2153/ac3ffb |
| [66] | OPENAI, ACHIAM J, ADLER S, et al. GPT-4 Technical Report[Z]. arXiv, 2024[2025-07-13]. http://arxiv.org/abs/2303.08774. |
| [67] | KEVIAN D, SYED U, GUO X, et al. Capabilities of Large Language Models in Control Engineering: A Benchmark Study on GPT-4, Claude 3 Opus, and Gemini 1.0 Ultra[Z]. arXiv, 2024[2025-07-13]. http://arxiv.org/abs/2404.03647. |
| [68] | TEAM G, ANIL R, BORGEAUD S, et al. Gemini: A Family of Highly Capable Multimodal Models[Z]. arXiv, 2025[2025-07-13]. http://arxiv.org/abs/2312.11805. |
| [69] |
FREY N C, SOKLASKI R, AXELROD S, et al. Neural scaling of deep chemical models[J]. Nature Machine Intelligence, 2023, 5(11): 1297-1305.
doi: 10.1038/s42256-023-00740-3 |
| [70] |
BAGAL V, AGGARWAL R, VINOD P K, et al. MolGPT: Molecular Generation Using a Transformer-Decoder Model[J]. Journal of Chemical Information and Modeling, 2022, 62(9): 2064-2076.
doi: 10.1021/acs.jcim.1c00600 |
| [71] | LI Y, GAO C, SONG X, et al. DrugGPT: A GPT-based Strategy for Designing Potential Ligands Targeting Specific Proteins[J]. bioRxiv, 2023: 2023.06.29.543848. |
| [72] | FANG Y, LIANG X, ZHANG N, et al. Mol-Instructions: A Large-Scale Biomolecular Instruction Dataset for Large Language Models[Z]. arXiv, 2024[2025-07-13]. http://arxiv.org/abs/2306.08018. |
| [73] | LUO Y, YANG K, HONG M, et al. MolFM: A Multimodal Molecular Foundation Model[Z]. arXiv, 2023[2025-07-13]. http://arxiv.org/abs/2307.09484. |
| [74] |
SEGLER M H S, KOGEJ T, TYRCHAN C, et al. Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks[J]. ACS Central Science, 2018, 4(1): 120-131.
doi: 10.1021/acscentsci.7b00512 pmid: 29392184 |
| [75] | HONDA S, SHI S, UEDA H R. SMILES Transformer: Pre-trained Molecular Fingerprint for Low Data Drug Discovery[Z]. arXiv, 2019[2025-07-13]. http://arxiv.org/abs/1911.04738. |
| [76] | SIMONOVSKY M, KOMODAKIS N. GraphVAE: Towards Generation of Small Graphs Using Variational Autoencoders[Z]. arXiv, 2018[2025-07-13]. http://arxiv.org/abs/1802.03480. |
| [77] | SHI C, XU M, ZHU Z, et al. GraphAF: a Flow-based Autoregressive Model for Molecular Graph Generation[Z]. arXiv, 2020[2025-07-13]. http://arxiv.org/abs/2001.09382. |
| [78] | XIANG Y, ZHAO H, MA C, et al. Instruction-Based Molecular Graph Generation with Unified Text-Graph Diffusion Model[Z]. arXiv, 2024[2025-07-21]. http://arxiv.org/abs/2408.09896. |
| [79] | NI Y, FENG S, CHI H, et al. Straight-Line Diffusion Model for Efficient 3D Molecular Generation[Z]. arXiv, 2025[2025-07-21]. http://arxiv.org/abs/2503.02918. |
| [80] | KIRCHMEYER M, PINHEIRO P O, SAREMI S. Score-based 3D molecule generation with neural fields[Z]. arXiv, 2025[2025-07-21]. http://arxiv.org/abs/2501.08508. |
| [81] |
ZHANG O, WANG T, WENG G, et al. Learning on topological surface and geometric structure for 3D molecular generation[J]. Nature Computational Science, 2023, 3(10): 849-859.
doi: 10.1038/s43588-023-00530-2 pmid: 38177756 |
| [82] |
ZHANG O, ZHANG J, JIN J, et al. ResGen is a pocket-aware 3D molecular generation model based on parallel multiscale modelling[J]. Nature Machine Intelligence, 2023, 5(9):1020-1030.
doi: 10.1038/s42256-023-00712-7 |
| [83] | SCHNEUING A, HARRIS C, DU Y, et al. Structure-based Drug Design with Equivariant Diffusion Models[Z]. arXiv, 2024[2025-07-13]. http://arxiv.org/abs/2210.13695. |
| [84] | GUAN J, QIAN W W, PENG X, et al. 3D Equivariant Diffusion for Target-Aware Molecule Generation and Affinity Prediction[Z]. arXiv, 2023[2025-07-13]. http://arxiv.org/abs/2303.03543. |
| [85] | GUAN J, ZHOU X, YANG Y, et al. DecompDiff: Diffusion Models with Decomposed Priors for Structure-Based Drug Design[Z]. arXiv, 2024[2025-07-13]. http://arxiv.org/abs/2403.07902. |
| [86] | QIAN H, HUANG W, TU S, et al. KGDiff: towards explainable target-aware molecule generation with knowledge guidance[J]. Briefings in Bioinformatics, 2023, 25(1): bbad435. |
| [87] |
DU H, JIANG D, ZHANG O, et al. A flexible data-free framework for structure-based de novo drug design with reinforcement learning[J]. Chemical Science, 2023, 14(43): 12166-12181.
doi: 10.1039/D3SC04091G |
| [88] |
ZHANG O, HUANG Y, CHENG S, et al. FragGen: towards 3D geometry reliable fragment-based molecular generation[J]. Chemical Science, 2024, 15(46): 19452-19465.
doi: 10.1039/d4sc04620j pmid: 39568888 |
| [89] | DIAO Y, HU F, SHEN Z, et al. MacFrag: segmenting large-scale molecules to obtain diverse fragments with high qualities[J]. Bioinformatics, 2023, 39(1): btad012. |
| [90] |
SYDOW D, SCHMIEL P, MORTIER J, et al. KinFragLib: Exploring the Kinase Inhibitor Space Using Subpocket-Focused Fragmentation and Recombination[J]. Journal of Chemical Information and Modeling, 2020, 60(12): 6081-6094.
doi: 10.1021/acs.jcim.0c00839 pmid: 33155465 |
| [91] | LEE S, KREIS K, VECCHAM S P, et al. Molecule Generation with Fragment Retrieval Augmentation[Z]. arXiv, 2024[2025-07-13]. http://arxiv.org/abs/2411.12078. |
| [92] |
IMRIE F, HADFIELD T E, BRADLEY A R, et al. Deep generative design with 3D pharmacophoric constraints[J]. Chemical Science, 2021, 12(43): 14577-14589.
doi: 10.1039/d1sc02436a pmid: 34881010 |
| [93] |
ZHU H, ZHOU R, CAO D, et al. A pharmacophore-guided deep learning approach for bioactive molecular generation[J]. Nature Communications, 2023, 14(1): 6234.
doi: 10.1038/s41467-023-41454-9 pmid: 37803000 |
| [94] | XIE W, ZHANG J, XIE Q, et al. Accelerating Discovery of Novel and Bioactive Ligands With Pharmacophore-Informed Generative Models[Z]. arXiv, 2024[2025-07-13]. http://arxiv.org/abs/2401.01059. |
| [95] | ALAKHDAR A, POCZOS B, WASHBURN N. Pharmacophore-Conditioned Diffusion Model for Ligand-Based De Novo Drug Design[Z]. arXiv, 2025[2025-07-21]. http://arxiv.org/abs/2505.10545. |
| [96] | SUN M, XING J, MENG H, et al. MolSearch: Search-based Multi-objective Molecular Generation and Property Optimization[C]// Proceedings of the 28th ACM SIGKDD Conference on Knowledge Discovery and Data Mining. Washington DC USA: ACM, 2022: 4724-4732. |
| [97] |
LIU Y, ZHU Y, WANG J, et al. A Multi-Objective Molecular Generation Method Based on Pareto Algorithm and Monte Carlo Tree Search[J]. Advanced Science, 2025, 12(20): 2410640.
doi: 10.1002/advs.v12.20 |
| [98] |
CHEN S. Structure-aware dual-target drug design through collaborative learning of pharmacophore combination and molecular simulation[J]. Chemical Science, 2024, 15(27): 10366-10380.
doi: 10.1039/d4sc00094c pmid: 38994407 |
| [99] |
BHATTACHARYA D, CASSADY H J, HICKNER M A, et al. Large Language Models as Molecular Design Engines[J]. Journal of Chemical Information and Modeling, 2024, 64(18): 7086-7096.
doi: 10.1021/acs.jcim.4c01396 pmid: 39231030 |
| [100] | ZENG Z, YIN B, WANG S, et al. Interactive Molecular Discovery with Natural Language[Z]. arXiv, 2023[2025-07-14]. http://arxiv.org/abs/2306.11976. |
| [101] |
ISHIDA S, SATO T, HONMA T, et al. Large language models open new way of AI-assisted molecule design for chemists[J]. Journal of Cheminformatics, 2025, 17(1): 36.
doi: 10.1186/s13321-025-00984-8 pmid: 40128788 |
| [102] | MALIKUSSAID, NUHA H H. VALID-Mol: a Systematic Framework for Validated LLM-Assisted Molecular Design[Z]. arXiv, 2025[2025-07-21]. http://arxiv.org/abs/2506.23339. |
| [103] |
MA P, CHENG Z, CHENG Z, et al. Discovery of EP4 antagonists with image-guided explainable deep learning workflow[J]. National Science Open, 2025, 4(4): 20240015.
doi: 10.1360/nso/20240015 |
| [104] | GAO B, HUANG Y, LIU Y, et al. PharmAgents: Building a Virtual Pharma with Large Language Model Agents[Z]. arXiv, 2025[2025-07-14]. http://arxiv.org/abs/2503.22164. |
| [105] |
BICKERTON G R, PAOLINI G V, BESNARD J, et al. Quantifying the chemical beauty of drugs[J]. Nature Chemistry, 2012, 4(2): 90-98.
doi: 10.1038/nchem.1243 pmid: 22270643 |
| [106] |
ERTL P, SCHUFFENHAUER A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions[J]. Journal of Cheminformatics, 2009, 1(1): 8.
doi: 10.1186/1758-2946-1-8 pmid: 20298526 |
| [107] |
TROTT O, OLSON A J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading[J]. Journal of computational chemistry, 2010, 31(2): 455-461.
doi: 10.1002/jcc.v31:2 |
| [108] |
FRIESNER R A, BANKS J L, MURPHY R B, et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy[J]. Journal of Medicinal Chemistry, 2004, 47(7): 1739-1749.
doi: 10.1021/jm0306430 |
| [109] |
SHEN C, ZHANG X, DENG Y, et al. Boosting Protein-Ligand Binding Pose Prediction and Virtual Screening Based on Residue-Atom Distance Likelihood Potential and Graph Transformer[J]. Journal of Medicinal Chemistry, 2022, 65(15): 10691-10706.
doi: 10.1021/acs.jmedchem.2c00991 pmid: 35917397 |
| [110] | ÖZTÜRK H, ÖZGÜR A, OZKIRIMLI E. DeepDTA: deep drug-target binding affinity prediction[J]. Bioinformatics, 2018, 34(17): i821-i829. |
| [111] |
DAINA A, MICHIELIN O, ZOETE V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules[J]. Scientific Reports, 2017, 7(1): 42717.
doi: 10.1038/srep42717 |
| [112] | BUTTENSCHOEN M, MORRIS G M, DEANE C M. PoseBusters: AI-based docking methods fail to generate physically valid poses or generalise to novel sequences[Z]. arXiv, 2023[2024-12-09]. http://arxiv.org/abs/2308.05777. |
| [1] | 何邦彦, 李琦, 孙哲南, 王蕊. 探索基于3D变换的后处理对3D对抗性点云迁移性的影响[J]. 数据与计算发展前沿, 2026, 8(2): 123-140. |
| [2] | 张耀南, 刘景琦, 康建芳, 南卓铜, 田文彪, 敏玉芳, 赵书萍, 王保得. 冰冻圈“大数据+AI+模型”耦合研究范式探索[J]. 数据与计算发展前沿, 2026, 8(2): 3-14. |
| [3] | 刘景琦, 张耀南, 康建芳, 刘杰, 杨治纬, 张智星, 王保得. 基于多智能体协同的高寒山区道路结冰数据工程预警[J]. 数据与计算发展前沿, 2026, 8(2): 40-53. |
| [4] | 张森, 褚铮, 黄松睿, 孟哲令. 中小学人工智能教育综述[J]. 数据与计算发展前沿, 2025, 7(6): 170-178. |
| [5] | 陈长松, 吴跃顺, 梅广. 人工智能安全防护体系的层次化模型研究[J]. 数据与计算发展前沿, 2025, 7(6): 68-76. |
| [6] | 郭惠婕,周泳杰,许建真. 基于多模态人工智能数据融合的中药药动学研究进展[J]. 数据与计算发展前沿, 2025, 7(2): 149-160. |
| [7] | 贾子昂. 基于多源半监督学习的牙齿结构分割[J]. 数据与计算发展前沿, 2025, 7(2): 175-185. |
| [8] | 蔡华谦, 刘逸豪, 关天鹏, 吴恺东, 杨婧如, 罗超然, 朱小杰, 刘佳, 黄罡. DPML: 一种面向科学数据语用的标记语言[J]. 数据与计算发展前沿, 2024, 6(4): 46-58. |
| [9] | 王志永, 刘晶晶, 王新明, 陈博文, 聂伟, 张瀚林, 刘洪海. 孤独症人工智能诊疗进展及前沿[J]. 数据与计算发展前沿, 2024, 6(3): 15-27. |
| [10] | 寇大治. 基于深度学习的口腔全景片牙齿自动分割方法[J]. 数据与计算发展前沿, 2024, 6(3): 162-172. |
| [11] | 何睿琳, 杨欣怡, 孙洪赞, 李晨. 基于图特征的组织病理学图像分析方法的最新发展情况与展望[J]. 数据与计算发展前沿, 2024, 6(2): 101-116. |
| [12] | 朱明明, 曹无敌, 吴林, 王自溪, 廖琦, 张思, 唐晓, 李杰, 王婧, 王彦棡, 王自发. 基于人工智能与大数据的双碳大气环境信息化应用进展与展望[J]. 数据与计算发展前沿, 2023, 5(3): 2-12. |
| [13] | 胡晓彦,徐寄遥,邹自明. “大数据&人工智能”驱动的空间天气科研范式变革初步探索[J]. 数据与计算发展前沿, 2023, 5(2): 24-36. |
| [14] | 齐法制,李刚,李纯,汪璐,张一,张正德,陈刚,罗武鸣,赵丽娜,胡誉,袁野. 基于人工智能的高能物理大数据技术与应用[J]. 数据与计算发展前沿, 2023, 5(2): 50-59. |
| [15] | 王凡,冯立强,曹荣强. 大数据驱动的海洋人工智能服务平台设计与应用[J]. 数据与计算发展前沿, 2023, 5(2): 73-85. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||